
生成式人工智慧(Generative Artificial Intelligence, GenAI)已廣泛應用於醫療領域,例如影像判讀、治療建議、疾病預測與病歷撰寫等。然而,GenAI醫療器材具備持續學習新資料的能力,模型本身也會隨之動態變化,可能讓結果變得難以預測,且難以重複驗證。一般而言,傳統機器學習醫療器材須透過嚴謹的臨床試驗,以驗證特定適應症的安全性與有效性,若欲擴增適應症,仍須進行額外的臨床評估。若針對GenAI醫療器材仍採用傳統審查方式,不僅難以因應其技術特性,亦可能導致審查流程冗長、成本高昂,進而限制創新發展。此外,GenAI導入臨床應用時,亦須審慎評估資料誤差與偏差風險、模型可解釋性的限制,以及結果一致性不足等挑戰。
為因應上述特性,韓國食品藥物安全處(Ministry of Food and Drug Safety, MFDS)於2025年1月24日發布全球首份《生成式人工智慧醫療器材之許可與審查指引》(??? ???? ???? ??·?? ????? (??? ???))。該指引針對GenAI醫療器材的特性,明確規範審查標準與作業程序,內容涵蓋適用範圍、產品範例、風險管理、申請文件撰寫、性能驗證及臨床有效性確認等項目,以提升審查透明度與申請便利性,並展現韓國於數位健康領域的政策前瞻與國際領導地位。
該指引適用於《數位醫療產品法》(??? ?????)第2條定義,目的為診斷、治療或預後的GenAI醫療器材,涵蓋製造、進口許可、認證以及臨床試驗等面向。至於醫療器材上市後管理、使用者(醫療機構等)的資安管理或不直接影響使用者健康的個人資料外洩等情形,則建議遵循《醫療法》(???)與《個人資料保護法》(???? ???)等其他相關法規。
該指引指出,GenAI醫療器材的判斷須依據其使用目的與生成模型的應用方式而定。若產品用途與形式難以明確歸類,建議向MFDS諮詢。舉例而言,屬於GenAI醫療器材的產品包括:依據胸部X光影像自動生成肺部病變報告、依據電子病歷(EMR)提供個別化治療建議、分析語音以早期發現巴金森氏症的工具,以及基因資料風險預測報告軟體;相對地,單純用於EMR記錄與查詢,或將語音轉為文字病歷的工具,則不屬於此範疇。此外,MFDS亦可依風險評估與現況調查結果,適時將具潛在風險的產品納入醫療器材管理,並透過指引修訂與利害關係人意見徵詢等程序加以調整。
無論是傳統醫療器材或GenAI醫療器材,都能透過ISO 14971:2019的風險管理流程,確認風險是否已降至可接受的範圍內。該指引依據GenAI醫療器材的特性,提出可能產生的風險因子(如下圖)。製造商可依據產品特性,識別相關風險因子,並採用ISO 14971:2019所訂程序進行風險管理。須注意,圖中所列風險因子未必皆適用於所有GenAI醫療器材,亦可能無法涵蓋全部潛在風險。
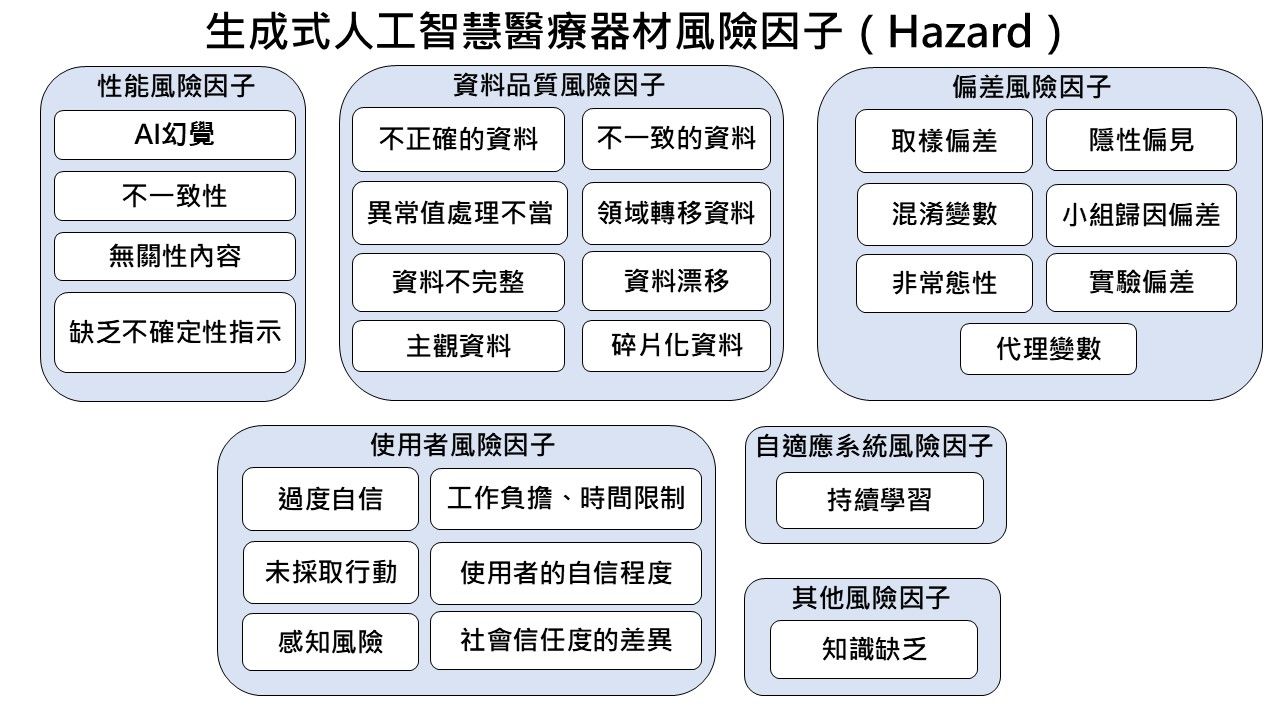 圖1、韓國指引說明製造商應識別生成式人工智慧醫療器材相關風險因子並依循ISO 14971:2019進行風險管理 (資料來源:資策會科法所整理自韓國食品藥物安全處(2025))
圖1、韓國指引說明製造商應識別生成式人工智慧醫療器材相關風險因子並依循ISO 14971:2019進行風險管理 (資料來源:資策會科法所整理自韓國食品藥物安全處(2025))
1. 填寫許可申請表
首先,申請者應依照《數位醫療產品許可、認證、申報、審查與評價等相關規定(??????? ??·??·??·?? ? ?? ?? ?? ??)》第10至21條之相關條文,填寫包括名稱、產品代碼和等級、外觀與結構、零組件或構成要素、製造流程、性能(功能)或特性、使用目的、如何使用、使用時注意事項、測試標準、製造商、儲存方法和使用期限等項目。在該指引中,考量到GenAI醫療器材的特性,對許可申請表中的適用項目進行調整,並提供相關範例進行說明。
例如,在「外觀與結構」項目中,需說明生成式AI模型的作用原理,並附上結構與資通訊系統圖;在「零組件或構成要素」部分,應記載AI模型運行所需的本地或雲端環境;而在「性能(功能)或特性」方面,則需說明模型輸入與輸出資訊、訓練資料來源及更新週期、雲端服務的類型及組成形式等內容;此外,「使用時注意事項」亦應明確揭示AI幻覺、性能漂移(Drift)及版本變動風險,並建議由臨床醫師審閱生成結果,確保安全性與有效性。
2. 分析性能驗證
GenAI醫療器材屬於《數位醫療產品法》(????????)規定的數位醫療器材軟體,須按照《數位醫療產品許可、認證、申報、審查與評價等相關規定(??????? ??·??·??·?? ? ?? ?? ?? ??)》附件1的要求,提交附件2格式的「數位醫療器材軟體適合性確認報告」與「軟體驗證和有效性確認資料」。相關資料的撰寫方式,可參閱《醫療器材軟體之許可與審查指引(申請人指引)》。此外,根據《數位醫療產品法實施規則(???????? ????)》,GenAI醫療器材必須繳交「使用適合性的相關資料」與「防止入侵他人電腦的保護措施之相關資料」。
指引另說明,申請人可依產品特性選用適當指標,評估生成結果的文字一致性與語意相似度,常見評估方法如下圖所示。
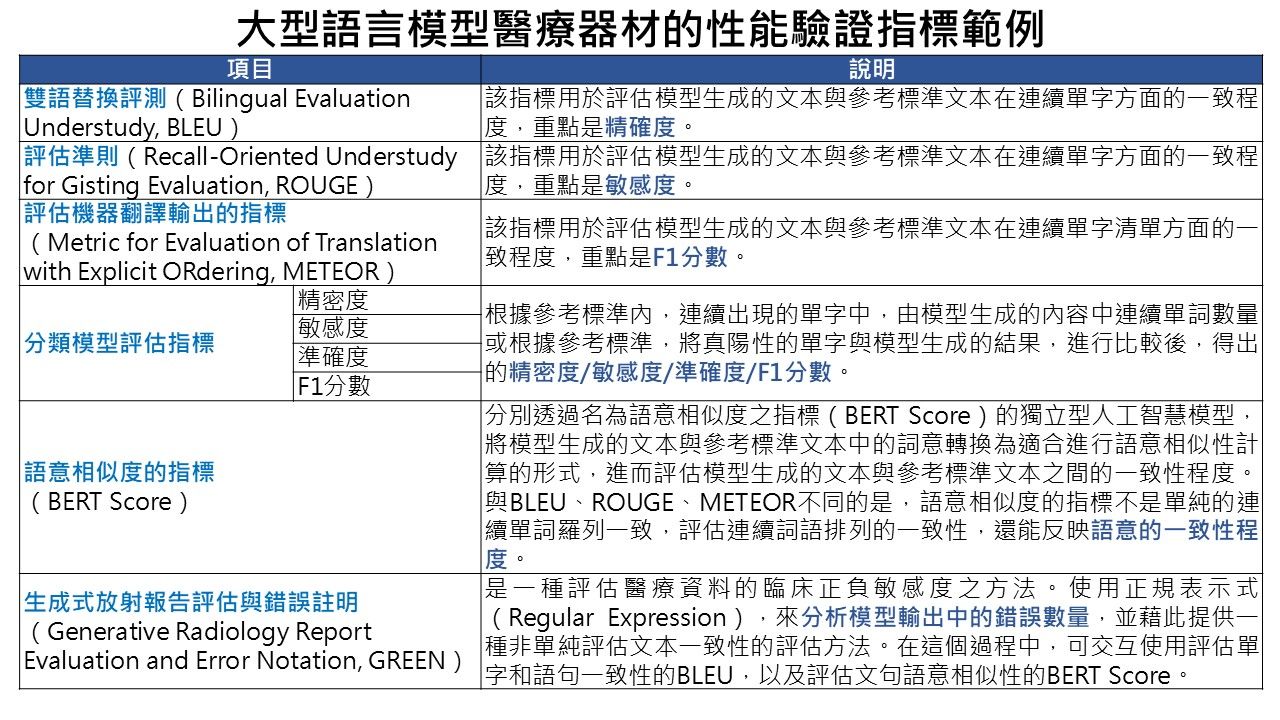
圖2、韓國指引提供生成式人工智慧醫療器材性能驗證指標範例 (資料來源:資策會科法所整理自韓國食品藥物安全處(2025))
3. 臨床有效性驗證
在臨床有效性驗證部分,指引建議可透過可透過醫療人員進行臨床評估(如五級評分法、RADPEER評分系統)或實際臨床試驗方式,評估生成內容的臨床意義與安全性,同時鼓勵在產品上市後,持續透過真實世界資料(RWD)建立真實世界證據(RWE),強化長期追蹤與風險管理。臨床評估方式如下圖所示。
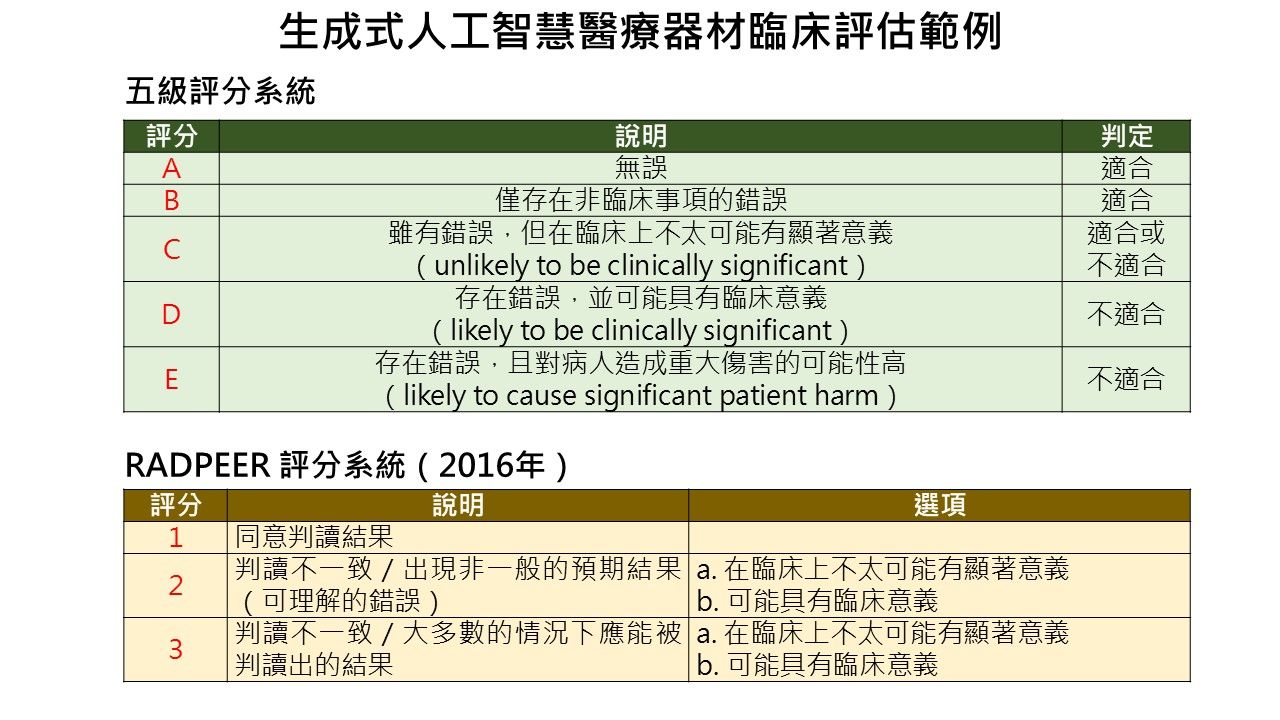
圖3、韓國指引提供生成式人工智慧醫療器材臨床評估範例 (資料來源:資策會科法所整理自韓國食品藥物安全處(2025))
韓國食品藥物安全處此次發布的《生成式人工智慧醫療器材之許可與審查指引》,不僅是全球首份針對GenAI醫療器材的專屬審查規範,也為各國主管機關提供了一套具體、前瞻且可操作的監管框架。該指引充分考量生成式AI技術在醫療應用上的創新潛力與風險特性,透過明確的審查程序、性能評估指標及臨床驗證方法,有效平衡安全性與促進創新之間的需求。對我國而言,面對AI技術於醫療器材領域的快速發展,如何建立具備透明性、風險管理與彈性調整機制的監管模式,將是推動AI生技產業發展與強化國際競爭力的重要課題。